 CCLE
CCLE
Cancer Proteomes.

The rumors are true! The Gygi Lab has a ton of exciting projects, from pre-clinical proteomics to in silico development projects.
Progress in science depends on new techniques, new discoveries and new ideas, probably in that order.
-Sydney Brenner
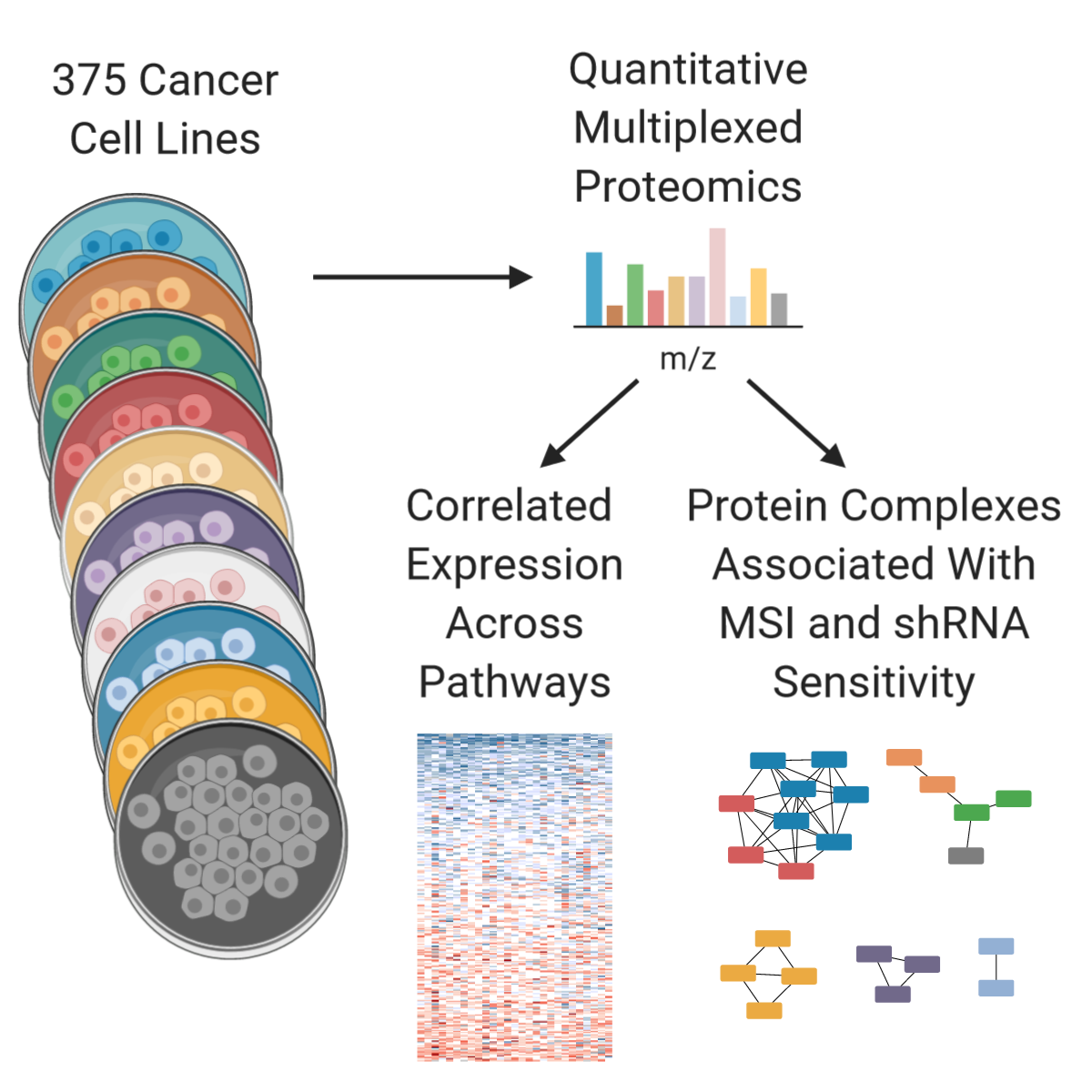
We recently expanded the Cancer Cell Line Encyclopedia to include quantitative proteome profiling by mass spectrometry of 375 cell lines from diverse lineages. The experiment was performed in multiplex format with 9 biological samples per plex and one common sample to normalize between plexes.
We quantify an average of over 9,000 proteins per experiment across 42 multiplex experiments consisting of 504 runs on the mass spectrometer and over 1,500 hours of instrument time. Additionally, the first two multiplexed experiments were performed in biological triplicate, the results of which are included here.
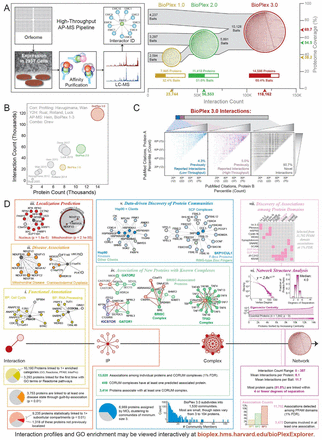
Thousands of interactions assemble proteins into modules that impart spatial and functional organization to the cellular proteome. Through affinity-purification mass spectrometry, we have created two proteome-scale, cell-line-specific interaction networks. The first, BioPlex 3.0, results from affinity purification of 10,128 human proteins – half the proteome – in 293T cells and includes 118,162 interactions among 14,586 proteins; the second results from 5,522 immunoprecipitations in HCT116 cells. These networks model the interactome at unprecedented scale, encoding protein function, localization, and complex membership. Their comparison validates thousands of interactions and reveals extensive customization of each network. While shared interactions reside in core complexes and involve essential proteins, cell-specific interactions bridge conserved complexes, likely ‘rewiring’ each cell’s interactome. Interactions are gained and lost in tandem among proteins of shared function as the proteome remodels to produce each cell’s phenotype. Viewable interactively online through BioPlexExplorer, these networks define principles of proteome organization and enable unknown protein characterization.
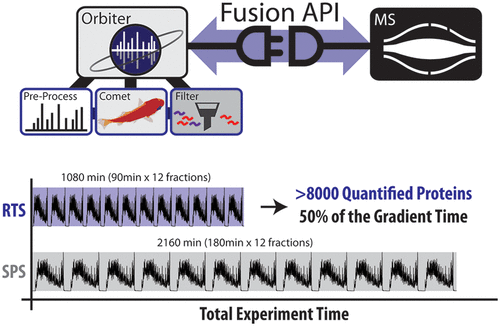
Multiplexed quantitative analyses of complex proteomes enable deep biological insight. While a multitude of workflows have been developed for multiplexed analyses, the most quantitatively accurate method (SPS-MS3) suffers from long acquisition duty cycles. We built a new, real-time database search (RTS) platform, Orbiter, to combat the SPS-MS3 method’s longer duty cycles. RTS with Orbiter eliminates SPS-MS3 scans if no peptide matches to a given spectrum. With Orbiter’s online proteomic analytical pipeline, which includes RTS and false discovery rate analysis, it was possible to process a single spectrum database search in less than 10 ms. The result is a fast, functional means to identify peptide spectral matches using Comet, filter these matches, and more efficiently quantify proteins of interest. Importantly, the use of Comet for peptide spectral matching allowed for a fully featured search, including analysis of post-translational modifications, with well-known and extensively validated scoring. These data could then be used to trigger subsequent scans in an adaptive and flexible manner. In this work we tested the utility of this adaptive data acquisition platform to improve the efficiency and accuracy of multiplexed quantitative experiments. We found that RTS enabled a 2-fold increase in mass spectrometric data acquisition efficiency. Orbiter’s RTS quantified more than 8000 proteins across 10 proteomes in half the time of an SPS-MS3 analysis (18 h for RTS, 36 h for SPS-MS3).
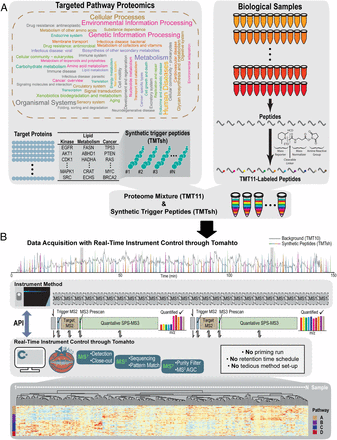
A user-friendly workflow, termed Tomahto, enables real-time targeted pathway proteomics assays using two-dimensional multiplexing technology. Hundreds of proteins of interest from a multitude of samples can be targeted and accurately quantified in a remarkably sensitive fashion. We highlight Tomahto’s ease of use, sensitivity, and accuracy and present proof-of-principle utility by targeting 260 metabolism- and inflammation-related proteins across 90 samples (nine tissues from five old and five young mice). Tissue-specific aging effects are presented with specific interest in metabolic and inflammatory pathways of white adipose tissue. We validated our approach through comparison with a global proteome survey across the tissues, work that we also provide as a general resource for the community.